Huntington Disease

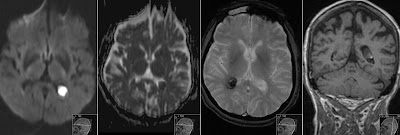
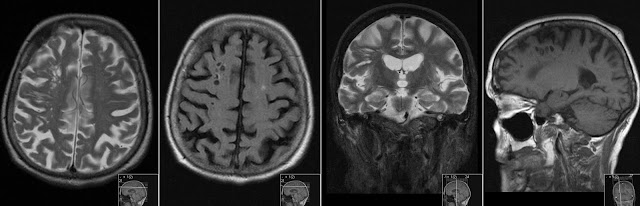
10 years old boy from parents being cousins to each other presents with progressive rigidity and dystonia. Initial MR showing significant bilateral atrophy in caudate nuclei and putamina with high signal on FLAIR and T2 consistent with gliosis.

Note striking symmetric atrophy of the putamina and caudate nuclei on T2 and IR.
Huntington disease (HD) is also known as Huntington chorea. HD is an autosomal dominant chronic hereditary neurodegenerative disorder with complete penetrance [Osborn].
Aggregates of huntingtin protein accumulate in axonal terminals, which eventually leads to the death of medium spiny neurons. Autopsy shows generalized cerebral atrophy with an average of 30% reduction in brain weight. Both the cortex and hemispheric WM are affected. The most characteristic gross abnormality is volume loss with rarefaction of the caudate nucleus, putamen, and globus pallidus [Osborn].
Microscopically, HD features neuronal loss with huntingtin nuclear inclusions, astrocytic gliosis, and iron accumulation. The changes are most severe in the basal ganglia but can also be seen in other regions of the brain, including the cerebellum [Osborn].
Juvenile-onset HD is initially characterized by rigidity and dystonia, much more than by chorea [Osborn].